By: Anne Child MD FRCP, Marjan Jahangiri FRCS (CTh), Alex Wan MRes, Genetics Research Centre and Cardiothoracic Surgery, St Georges University of London and St Georges NHS Hospitals Foundation Trust
Aortopathy (a″or-top´ah-thē) simply means any disease of the aorta, the largest blood vessel to leave the heart, carrying oxygenated blood to every part of the body. Disease of the aorta within the chest is called thoracic aortic disease. We are focused on thoracic aortic aneurysms (enlargement) and dissection (tearing), especially the 20% of cases which are heritable, passed directly from parent to child with a 50-50 risk of involvement whether male or female.
Aortopathy is of two types; thoracic aortic enlargement progressing to the life-threatening complication of acute aortic dissection; or thoracic aortic dissection in the absence of aortic enlargement.
In some instances, the aortic involvement is part of a collection of physical signs and symptoms, known as a syndrome. The associated features help the physician or surgeon to make a diagnosis of the underlying cause. For example, Marfan syndrome is familial, and associated with characteristic findings in the skeletal system (tall and thin), the eye (dislocated eye lenses), and the heart (aortic aneurysm and dissection). In this syndrome, the gene for the disorder has already been discovered. Close family members (siblings, parents, children) of the affected person may then be screened by DNA testing to see if they carry the predisposing gene. This allows the doctor to decide if the patient needs yearly ultrasound pictures of the heart (echocardiogram) or other screening method (MRI= magnetic resonance imaging) or CT scanning (computed tomography of the chest). Medication is prescribed to delay the enlargement of the aorta if progression is shown from year to year.
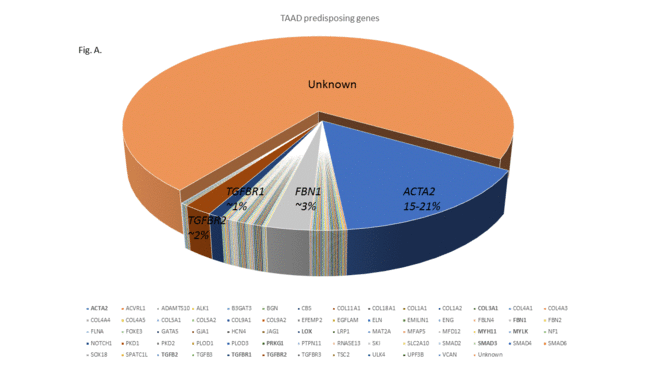
At present, over 50 genes are known to predispose to aortic aneurysms, and research adds to this list annually. These genes cause the protein constituents of the aortic wall to be produced in less than normal quantities, weakening the wall, and allowing expansion and tearing. This research will lead to better understanding of aortopathy, and future preventive medication or gene therapy.
The diagram in Fig.A below indicates all the genes at present known to predispose, in a pie diagram. Some genes are major, and some only contribute 1 to 2%, and are rare.
With each gene, there may be physical features in the patient which lead to a diagnosis clinically of the underlying cause, such as early osteoarthritis of the knee, or very stretchy skin. Diagnosis is made on the basis of a detailed family history of similar conditions, physical examination of the patient, and a screening DNA test called an aortic disease panel. This is available through the NHS, when the patient is seen in a regional genetics unit.
Family DNA studies, and the 100K genome project are running in parallel to define new genes in families where no known gene is identified.
The condition is entirely treatable, but at present no cure is available except expert thoracic aortic surgery when the aortic root measures between 4.5 cm and 5 cm in diameter just above the aortic valve, in the sinus of Valsalva. In expert hands, such surgery is safe, with a 1 to 3% mortality risk, and thus a 97 to 99% chance of success. Replacement surgery adds at present 20 to 25 years of life, often extending the predicted lifespan into the normal range (70 to 80 years).
After surgery, annual full length scans of the aorta are recommended, since a small proportion of patients go on to develop a second or third aneurysm, again treatable surgically.
Two main types of surgery have become available. The most common recommended approach is to replace the defective aortic wall with open heart surgery. This should be performed by a surgeon expert in the technique, and there are several expert centres in the UK.
The second more experimental approach is to wrap the early part of the aorta with a mesh which becomes incorporated in the wall with growth. This procedure is called PEARS and is only offered in a few centres across Europe. Other forms of cardiac surgery may involve mending or replacing the aortic or mitral valves. Heart palpitations (arrhythmias) can also affect aortopathy patients, and once these are defined by electrocardiogram, and other studies, treatment with medication, a pacemaker or ablation surgery can be offered.
Genetic errors discovered in genes predisposing to aortopathy are grouped into four major categories based on international evidence of similar gene errors in patients. The gene is scored according to whether the evidence is strong, or weak. Patients presenting with isolated aneurysm and no other clinical features are less likely to demonstrate a gene mutation, but just to be safe, are kept under review, and their close relatives are recommended to have echocardiogram screening for aortic aneurysm.
Future Research Funding is required to speed the finding of all the genes within the next 10 years. Databases of mutations in genes already discovered need financial support.
For each gene discovered, protein studies must be performed to indicate how the new gene causes aortic wall weakening. Surgical research concentrates on the efficacy and safety of techniques at present being used, and improvement in life span.
In addition, education and awareness programmes to raise awareness among the public and physicians alike, is required.
Funding for our research programme would be very welcome. To donate, please click here.